


When it comes to medical device development, innovation gets all the excitement and attention, but the less glamorous regulatory process is a non-negotiable hurdle that can delay even the most advanced technology from reaching the market. That’s why it’s essential to establish a solid regulatory strategy that anticipates and mitigates risks, accelerates approvals, and ultimately aligns with your business goals, right from the start.
Kaleidoscope Innovation’s team of seasoned regulatory experts have developed an end-to-end regulatory assessment designed to map out the path to clearance or approval with efficiency and precision. Here’s an overview of our approach.
01
Background & Purpose: Laying the Foundation
The first step is to develop a comprehensive understanding of your product’s intended use, target markets, and previous regulatory history. It’s also essential to align your goals with regional requirements—whether for the U.S. (FDA), EU (MDR), or other global territories—ensuring that every decision is guided by both compliance and market strategy. Here are some guidelines we use:
- Define the Intended Use/Indications for Use
- Identify key territories for regulatory clearance
- Summarize any prior submissions, rejections, or correspondence to avoid duplication and leverage existing work
02
Product Description: Translating Innovation into Regulatory Language
Next, we break down your device’s design and functionality into a format that regulators understand, capturing the critical elements—from technical schematics to visual documentation—that describe the full scope of your product. This is an area in which Kaleidoscope excels. Our product assessments include:
- Detailed device description and illustrations
- Identification of any planned iterations that could impact classification or approval timelines
03
Medical Device Definition: Clarifying Classification Early
Accurate classification is critical, identifying whether your product meets the FDA’s definition of a medical device or qualifies under non-device pathways. These include:
- General Wellness classifications
- Non-device Clinical Decision Support (CDS) tools
By making this determination early, we help uncover faster or lower-risk pathways and avoid unnecessary regulatory burden.
04
Product Regulation: Finding the Right Regulatory Fit
Using FDA databases and internal expertise allows us to determine the appropriate regulatory classification and product codes based on the intended use and technology profile. Kaleidoscope’s regulatory specialists:
- Cross-reference public databases to identify potential regulations
- Select the most appropriate product codes and submission routes, whether a 510(k), De Novo, or PMA
05
Identifying Predicate & Reference Devices: Building a Case for Equivalence
If pursuing a 510(k), our team will research potential predicate devices to demonstrate substantial equivalence and streamline your path to market.
We also explore:
- Reference devices that can support safety and performance claims
- Strategies for leveraging this data to shorten timelines and reduce required testing
06
Regulatory Recommendations: Strategic Pathways to Market
This is not an area you can check boxes, and having an experienced team lead the effort eases the burden and stress for our clients. We craft a regulatory roadmap that aligns with your product and your business. Our process addresses:
- Early FDA engagement (Q-Sub, Pre-Sub, or Pre-IDE meetings), including communications and strategic negotiations that facilitate the best outcomes
- Opportunities to apply for Breakthrough Device Designation or Safer Technologies Program (STeP)
- The most efficient marketing pathway, such as a Special 510(k) or De Novo, based on cost, timing, and risk
07
Testing Requirements: Designing for Approval
Aligning our experience with your product needs, we develop a targeted testing strategy using:
- Relevant regulatory guidance documents
- Precedent data from similar cleared devices
- Listings on ClinicalTrials.gov
- Industry standards and FDA expectations
Our recommendations consider everything from biocompatibility and electrical safety to usability testing and human factors engineering (HFE). Our team can also go beyond testing to implementation, tapping Kaleidoscope’s in-house Human Factors team and/or wet lab, conducting clinical trials, and more.
08
Marketing Submission Content: Getting Everything in Order
We ensure you're fully prepared for submission with a checklist that covers every requirement. Our regulatory team compiles, reviews, and often authors the following, delivering final approved versions for company submission:
- All necessary 510(k), De Novo, or PMA documentation
- Establishment registration and device listing
- Small business qualification options, when applicable
- Quality System Regulation (QSR) readiness; we can also build your QMS from scratch in paper or digital format
- References to the most current FDA guidance documents and ISO standards
With decades of experience working with industry leaders like Baxter and a range of small start-up businesses, Kaleidoscope Innovation has earned a reputation for combining deep regulatory knowledge with world-class product development. Our approach helps new projects get started from the ground up and provides clearance for those that are further along. In all cases, we target long-term success, risk mitigation, and accelerated timelines.
Let’s map your regulatory path with confidence. > Let's start something.
Download this Regulatory Strategy At-a-Glance Guide to see how we simplify FDA submissions and accelerate approvals.
Back to Insights + News-
 |
|With over 25 years of experience in Regulatory Affairs, Colleen brings deep expertise in global medical device compliance, clinical trial strategy, and FDA, Health Canada, and ICH regulations—guiding products from protocol development through submissions, audits, and post-market support.
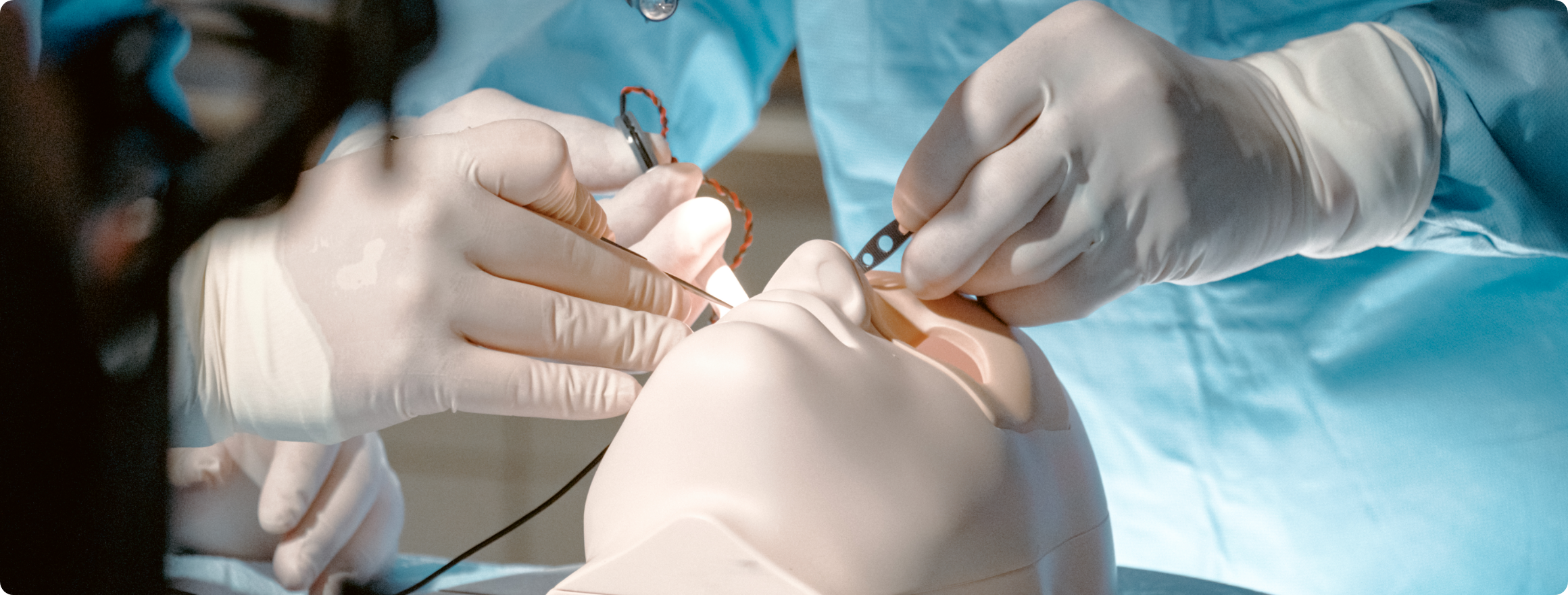

 Design and Prototyping
Design and Prototyping Formative Usability Testing and Evaluation
Formative Usability Testing and Evaluation

